



“Genome analysis reveals that bats may be the source of SARS-Cov-2.”
Christos Lynteris has drawn attention to the fundamental role of the “zoonotic diagram” in both conceptualizing and communicating the science of zoonosis. These diagrams provide a visual representation of the trajectory of pathogen transmission from one or more animal species to humans. For instance, typical diagrams of sylvatic plague use arrows or pathway lines (representing routes of pathogen transmission) to link rodents, fleas, and humans. Through a panoptic representation of the relationships of animals and humans in terms of lines of transmission, the zoonotic diagram prioritizes the prevention of human infection, pathologizes animals as sources of disease, and targets public health interventions at the animal–human interface.
Underlying and preceding the zoonotic diagram, other scientific visualizations articulate narratives of zoonotic emergence in more hypothetical modes, crafting stories about how a pathogen may have “spilled over” from animals to humans. Today, the most dominant among these is the phylogeny, or phylogenetic tree. A product of the modern evolutionary synthesis, phylogenetics is an approach to understanding the relationships among or within groups of organisms by situating them within genetic, evolutionary time. By comparing the whole genome sequences of two organisms (e.g. humans and chimpanzees, or two virus samples), researchers can calculate genomic variation and use this difference to infer the degree of relatedness on an evolutionary tree.
Immediately after the cluster of severe pneumonia cases was identified in Wuhan in December 2019, the Wuhan Institute of Virology sequenced the genome of the unknown new virus and compared the genome sequence with a range of sequences from other coronaviruses sampled and stored in the laboratory archive. The Institute reported that the novel SARS-CoV-2 virus shared only around 70% of its genome with the 2003 SARS-CoV virus, and instead most closely resembled a coronavirus previously isolated from a sample of bat feces taken from a cave in Yunnan Province, a virus referred to as RaTG-13. Using a range of software techniques, the lab then converted this similarity plot analysis into a phylogenetic tree to show that RaTG-13 is “the closest relative of [SARS-CoV-2] and they form a distinct lineage from other SARS-CoVs”. Based on this “close phylogenetic relationship”, the authors concluded that SARS-CoV-2 had a “probable bat origin” (See Figure 1).
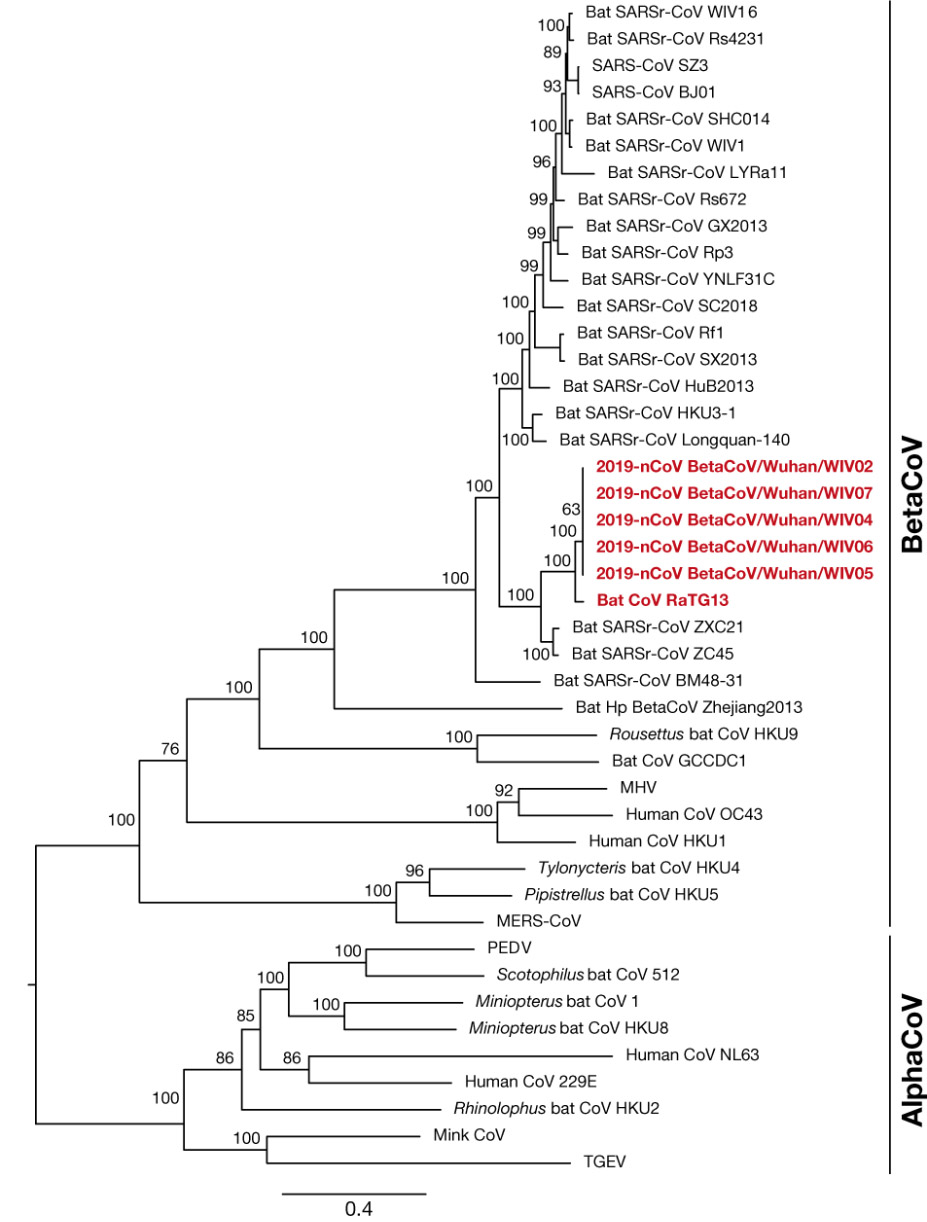
Phylogenetic tree showing “close phylogenetic relationship” of RaTG13 and SARS-CoV-2 (referred to at the time as 2019-nCoV). Source: Zhou et al 2020. Creative Commons 4.0.
As Stefan Helmreich has argued, phylogenetic analysis relies on the visual language of the family tree, that 19th century European diagram of animal breeding and family kinship, rooted in the logic of sex and descent. Yet viruses, including coronaviruses, are known to transfer genes horizontally through recombination when at least two viruses co-infect the same host cell. Helmreich argues that the frequency of recombination and its impact on viral evolution threatens to destabilize the utility of phylogenetic trees, as logics of descent and random mutation are replaced by models of gene “transfer”. When scientists locate virus samples within a branching phylogeny or calculate the date of evolutionary divergence from the most recent common ancestor, assumptions must be made not only about rates of mutation (which may be more or less stable) but also about the frequency and impact of recombination events. As Helmreich suggests regarding deep-sea microbes, the logic of recombination raises questions about whether phylogenetic trees of coronavirus genomes should be taken as narratives of how SARS-CoV-2 actually evolved.
Perhaps more concerning, however, is another feature of the way phylogenetic trees are used in the search for the origins of zoonotic viruses such as SARS-CoV-2: the fact that a comparison between two virus genomes (RaTG-13 and SARS-CoV-2) is used not only to infer an evolutionary relationship between the two viruses, but also taken to indicate a historical relationship of contact between two host species (bats and humans). Genomic variation stands for evolutionary divergence, which in turn is seen as evidence for interspecies contact and “spillover”. For example, the recent World Health Organization report on the Origins of SARS-CoV-2 reiterates the claim that bats are the probable source of the coronavirus, based almost exclusively on evidence from comparative genomics.
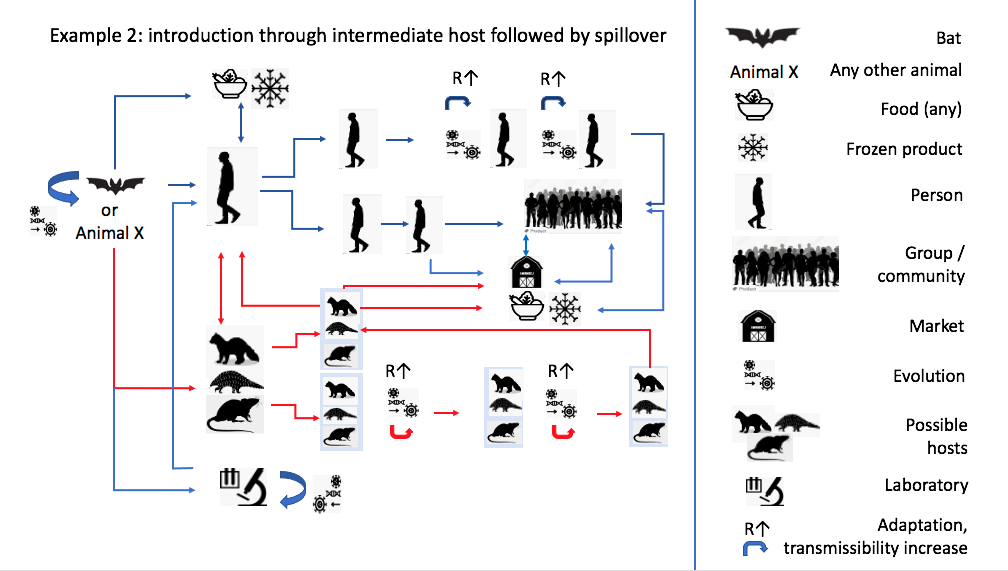
Zoonotic diagram based on phylogenetic inference. Source: Joint WHO–China Study, 2021.
The WHO report goes on to suggest that direct spillover from bats is less likely than introduction through an intermediate host (such as pangolins or mustelids). Still, the basis for this claim remains in the genomic register, either by citing the isolation of coronaviruses from other species that also closely resemble SARS-CoV-2 at the genomic level, or simply through the evolutionary logic itself: “Although the closest related viruses have been found in bats, the evolutionary distance between these bat viruses and SARS-CoV-2 is estimated to be several decades, suggesting a missing link”. It is particularly striking that the concept of “missing link”, an outdated term in evolutionary theory held over from the idea of the Great Chain of Being, is used here in the place usually occupied by terms like “bridge species” or “intermediate host” in studies of zoonotic transmission. Evolutionary inference is transposed into the form of a zoonotic diagram, and molecular resemblance between virus genomes comes to visually represent hypothetical lines of transmission between host species (Figure 2). Unfortunately, by searching for origins only in the branches of the viral family tree, we lose sight of other zones of virulence: the contemporary assemblages of humans and animals, landscapes and microbes that shape how viruses evolve, infect, recombine, and sometimes go pandemic.